Drug development is a complex and long process that involves screening thousands of drug candidates and using computational or experimental methods to evaluate leads. According to McKinsey, a single drug can take 10 years and cost an average of $2.6 billion to go through disease target identification, drug screening, drug-target validation, and eventual commercial launch. Drug discovery is the research component of this pipeline that generates candidate drugs with the highest likelihood of being effective with the least harm to patients. Machine learning (ML) methods can help identify suitable compounds at each stage in the drug discovery process, resulting in more streamlined drug prioritization and testing, saving billions in drug development costs (for more information, refer to AI in biopharma research: A time to focus and scale).
Drug targets are typically biological entities called proteins, the building blocks of life. The 3D structure of a protein determines how it interacts with a drug compound; therefore, understanding the protein 3D structure can add significant improvements to the drug development process by screening for drug compounds that fit the target protein structure better. Another area where protein structure prediction can be useful is understanding the diversity of proteins, so that we only select for drugs that selectively target specific proteins without affecting other proteins in the body (for more information, refer to Improving target assessment in biomedical research: the GOT-IT recommendations). Precise 3D structures of target proteins can enable drug design with higher specificity and lower likelihood of cross-interactions with other proteins.
However, predicting how proteins fold into their 3D structure is a difficult problem, and traditional experimental methods such as X-ray crystallography and NMR spectroscopy can be time-consuming and expensive. Recent advances in deep learning methods for protein research have shown promise in using neural networks to predict protein folding with remarkable accuracy. Folding algorithms like AlphaFold2, ESMFold, OpenFold, and RoseTTAFold can be used to quickly build accurate models of protein structures. Unfortunately, these models are computationally expensive to run and the results can be cumbersome to compare at the scale of thousands of candidate protein structures. A scalable solution for using these various tools will allow researchers and commercial R&D teams to quickly incorporate the latest advances in protein structure prediction, manage their experimentation processes, and collaborate with research partners.
Amazon SageMaker is a fully managed service to prepare, build, train, and deploy high-quality ML models quickly by bringing together a broad set of capabilities purpose-built for ML. It offers a fully managed environment for ML, abstracting away the infrastructure, data management, and scalability requirements so you can focus on building, training, and testing your models.
In this post, we present a fully managed ML solution with SageMaker that simplifies the operation of protein folding structure prediction workflows. We first discuss the solution at the high level and its user experience. Next, we walk you through how to easily set up compute-optimized workflows of AlphaFold2 and OpenFold with SageMaker. Finally, we demonstrate how you can track and compare protein structure predictions as part of a typical analysis. The code for this solution is available in the following GitHub repository.
Solution overview
In this solution, scientists can interactively launch protein folding experiments, analyze the 3D structure, monitor the job progress, and track the experiments in Amazon SageMaker Studio.
The following screenshot shows a single run of a protein folding workflow with Amazon SageMaker Studio. It includes the visualization of the 3D structure in a notebook, run status of the SageMaker jobs in the workflow, and links to the input parameters and output data and logs.

The following diagram illustrates the high-level solution architecture.

To understand the architecture, we first define the key components of a protein folding experiment as follows:
- FASTA target sequence file – The FASTA format is a text-based format for representing either nucleotide sequences or amino acid (protein) sequences, in which nucleotides or amino acids are represented using single-letter codes.
- Genetic databases – A genetic database is one or more sets of genetic data stored together with software to enable users to retrieve genetic data. Several genetic databases are required to run AlphaFold and OpenFold algorithms, such as BFD, MGnify, PDB70, PDB, PDB seqres, UniRef30 (FKA UniClust30), UniProt, and UniRef90.
- Multiple sequence alignment (MSA) – A sequence alignment is a way of arranging the primary sequences of a protein to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships between the sequences. The input features for predictions include MSA data.
- Protein structure prediction – The structure of input target sequences is predicted with folding algorithms like AlphaFold2 and OpenFold that use a multitrack transformer architecture trained on known protein templates.
- Visualization and metrics – Visualize the 3D structure with the py3Dmol library as an interactive 3D visualization. You can use metrics to evaluate and compare structure predictions, most notably root-mean-square deviation (RMSD) and template modeling Score (TM-score)
The workflow contains the following steps:
- Scientists use the web-based SageMaker ML IDE to explore the code base, build protein sequence analysis workflows in SageMaker Studio notebooks, and run protein folding pipelines via the graphical user interface in SageMaker Studio or the SageMaker SDK.
- Genetic and structure databases required by AlphaFold and OpenFold are downloaded prior to pipeline setup using Amazon SageMaker Processing, an ephemeral compute feature for ML data processing, to an Amazon Simple Storage Service (Amazon S3) bucket. With SageMaker Processing, you can run a long-running job with a proper compute without setting up any compute cluster and storage and without needing to shut down the cluster. Data is automatically saved to a specified S3 bucket location.
- An Amazon FSx for Lustre file system is set up, with the data repository being the S3 bucket location where the databases are saved. FSx for Lustre can scale to hundreds of GB/s of throughput and millions of IOPS with low-latency file retrieval. When starting an estimator job, SageMaker mounts the FSx for Lustre file system to the instance file system, then starts the script.
- Amazon SageMaker Pipelines is used to orchestrate multiple runs of protein folding algorithms. SageMaker Pipelines offers a desired visual interface for interactive job submission, traceability of the progress, and repeatability.
- Within a pipeline, two computationally heavy protein folding algorithms—AlphaFold and OpenFold—are run with SageMaker estimators. This configuration supports mounting of an FSx for Lustre file system for high throughput database search in the algorithms. A single inference run is divided into two steps: an MSA construction step using an optimal CPU instance and a structure prediction step using a GPU instance. These substeps, like SageMaker Processing in Step 2, are ephemeral, on-demand, and fully managed. Job output such as MSA files, predicted pdb structure files, and other metadata files are saved in a specified S3 location. A pipeline can be designed to run one single protein folding algorithm or run both AlphaFold and OpenFold after a common MSA construction.
- Runs of the protein folding prediction are automatically tracked by Amazon SageMaker Experiments for further analysis and comparison. The job logs are kept in Amazon CloudWatch for monitoring.
Prerequisites
To follow this post and run this solution, you need to have completed several prerequisites. Refer to the GitHub repository for a detailed explanation of each step.
- A SageMaker domain and a user profile – If you don’t have a SageMaker Studio domain, refer to Onboard to Amazon SageMaker Domain Using Quick Setup.
- IAM policies – Your user should have the AWS Identity and Access Management (IAM) AmazonSageMakerFullAccess policy attached, the ability to build Docker container images to Amazon Elastic Container Registry (Amazon ECR), and FSx for Lustre file systems created. See the readme for more details.
- Network – A VPC with an Amazon S3 VPC endpoint. We use this VPC location to provision the FSx for Lustre file system and SageMaker jobs.
- Docker resources – Run
00-prerequisite.ipynbfrom the repository to build the Docker images, download the genetic database to Amazon S3, and create an FSx for Lustre file system with a data repository association to the S3 bucket.
Run protein folding on SageMaker
We use the fully managed capabilities of SageMaker to run computationally heavy protein folding jobs without much infrastructure overhead. SageMaker uses container images to run custom scripts for generic data processing, training, and hosting. You can easily start an ephemeral job on-demand that runs a program with a container image with a couple of lines of the SageMaker SDK without self-managing any compute infrastructure. Specifically, the SageMaker estimator job provides flexibility when it comes to choice of container image, run script, and instance configuration, and supports a wide variety of storage options, including file systems such as FSx for Lustre. The following diagram illustrates this architecture.

Folding algorithms like AlphaFold and OpenFold use a multitrack transformer architecture trained on known protein templates to predict the structure of unknown peptide sequences. These predictions can be run on GPU instances to provide best throughput and lowest latency. The input features however for these predictions include MSA data. MSA algorithms are CPU-dependent and can require several hours of processing time.
Running both the MSA and structure prediction steps in the same computing environment can be cost-inefficient because the expensive GPU resources remain idle while the MSA step runs. Therefore, we optimize the workflow into two steps. First, we run a SageMaker estimator job on a CPU instance specifically to compute MSA alignment given a particular FASTA input sequence and source genetic databases. Then we run a SageMaker estimator job on a GPU instance to predict the protein structure with a given input MSA alignment and a folding algorithm like AlphaFold or OpenFold.
Run MSA generation
For MSA computation, we include a custom script run_create_alignment.sh and create_alignments.py script that is adopted from the existing AlphaFold prediction source run_alphafold.py. Note that this script may need to be updated if the source AlphaFold code is updated. The custom script is provided to the SageMaker estimator via script mode. The key components of the container image, script mode implementation, and setting up a SageMaker estimator job are also part of the next step of running folding algorithms, and are described further in the following section.
Run AlphaFold
We get started by running an AlphaFold structure prediction with a single protein sequence using SageMaker. Running an AlphaFold job involves three simple steps, as can be seen in 01-run_stepbystep.ipynb. First, we build a Docker container image based on AlphaFold’s Dockerfile so that we can also run AlphaFold in SageMaker. Second, we construct the script run_alphafold.sh that instructs how AlphaFold should be run. Third, we construct and run a SageMaker estimator with the script, the container, instance type, data, and configuration for the job.
Container image
The runtime requirement for a container image to run AlphaFold (OpenFold as well) in SageMaker can be greatly simplified with AlphaFold’s Dockerfile. We only need to add a handful of simple layers on top to install a SageMaker-specific Python library so that a SageMaker job can communicate with the container image. See the following code:
Input script
We then provide the script run_alphafold.sh that runs run_alphafold.py from the AlphaFold repository that is currently placed in the container /app/alphafold/run_alphafold.py. When this script is run, the location of the genetic databases and the input FASTA sequence will be populated by SageMaker as environment variables (SM_CHANNEL_GENETIC and SM_CHANNEL_FASTA, respectively). For more information, refer to Input Data Configuration.
Estimator job
We next create a job using a SageMaker estimator with the following key input arguments, which instruct SageMaker to run a specific script using a specified container with the instance type or count, your networking option of choice, and other parameters for the job. vpc_subnet_ids and security_group_ids instruct the job to run inside a specific VPC where the FSx for Lustre file system is in so that we can mount and access the filesystem in the SageMaker job. The output path refers to a S3 bucket location where the final product of AlphaFold will be uploaded to at the end of a successful job by SageMaker automatically. Here we also set a parameter DB_PRESET, for example, to be passed in and accessed within run_alphafold.sh as an environmental variable during runtime. See the following code:
from sagemaker.estimator import Estimator
alphafold_image_uri=f'{account}.dkr.ecr.{region}.amazonaws.com/sagemaker-studio-alphafold:v2.3.0'
instance_type='ml.g5.2xlarge'
instance_count=1
vpc_subnet_ids=['subnet-xxxxxxxxx'] # okay to use a default VPC
security_group_ids=['sg-xxxxxxxxx']
env={'DB_PRESET': db_preset} # <full_dbs|reduced_dbs>
output_path='s3://%s/%s/job-output/'%(default_bucket, prefix)
estimator_alphafold = Estimator(
source_dir='src', # directory where run_alphafold.sh and other runtime files locate
entry_point='run_alphafold.sh', # our script that runs /app/alphafold/run_alphafold.py
image_uri=alphafold_image_uri, # container image to use
instance_count=instance_count, #
instance_type=instance_type,
subnets=vpc_subnet_ids,
security_group_ids=security_group_ids,
environment=env,
output_path=output_path,
...)Finally, we gather the data and let the job know where they are. The fasta data channel is defined as an S3 data input that will be downloaded from an S3 location into the compute instance at the beginning of the job. This allows great flexibility to manage and specify the input sequence. On the other hand, the genetic data channel is defined as a FileSystemInput that will be mounted onto the instance at the beginning of the job. The use of an FSx for Lustre file system as a way to bring in close to 3 TB of data avoids repeatedly downloading data from an S3 bucket to a compute instance. We call the .fit method to kick off an AlphaFold job:
from sagemaker.inputs import FileSystemInput
file_system_id='fs-xxxxxxxxx'
fsx_mount_id='xxxxxxxx'
file_system_directory_path=f'/{fsx_mount_id}/{prefix}/alphafold-genetic-db' # should be the full prefix from the S3 data repository
file_system_access_mode='ro' # Specify the access mode (read-only)
file_system_type='FSxLustre' # Specify your file system type
genetic_db = FileSystemInput(
file_system_id=file_system_id,
file_system_type=file_system_type,
directory_path=file_system_directory_path,
file_system_access_mode=file_system_access_mode)
s3_fasta=sess.upload_data(path='sequence_input/T1030.fasta', # FASTA location locally
key_prefix='alphafoldv2/sequence_input') # S3 prefix. Bucket is sagemaker default bucket
fasta = sagemaker.inputs.TrainingInput(s3_fasta,
distribution='FullyReplicated',
s3_data_type='S3Prefix',
input_mode='File')
data_channels_alphafold = {'genetic': genetic_db, 'fasta': fasta}
estimator_alphafold.fit(inputs=data_channels_alphafold,
wait=False) # wait=False gets the cell back in the notebook; set to True to see the logs as the job progressesThat’s it. We just submitted a job to SageMaker to run AlphaFold. The logs and output including .pdb prediction files will be written to Amazon S3.
Run OpenFold
Running OpenFold in SageMaker follows a similar pattern, as shown in the second half of 01-run_stepbystep.ipynb. We first add a simple layer to get the SageMaker-specific library to make the container image SageMaker compatible on top of OpenFold’s Dockerfile. Secondly, we construct a run_openfold.sh as an entry point for the SageMaker job. In run_openfold.sh, we run the run_pretrained_openfold.py from OpenFold, which is available in the container image with the same genetic databases we downloaded for AlphaFold and OpenFold’s model weights (--openfold_checkpoint_path). In terms of input data locations, besides the genetic databases channel and the FASTA channel, we introduce a third channel, SM_CHANNEL_PARAM, so that we can flexibly pass in the model weights of choice from the estimator construct when we define and submit a job. With the SageMaker estimator, we can easily submit jobs with different entry_point, image_uri, environment, inputs, and other configurations for OpenFold with the same signature. For the data channel, we add a new channel, param, as an Amazon S3 input along with the use of the same genetic databases from the FSx for Lustre file system and FASTA file from Amazon S3. This, again, allows us easily specify the model weight to use from the job construct. See the following code:
s3_param=sess.upload_data(path='openfold_params/finetuning_ptm_2.pt',
key_prefix=f'{prefix}/openfold_params')
param = sagemaker.inputs.TrainingInput(s3_param,
distribution="FullyReplicated",
s3_data_type="S3Prefix",
input_mode='File')
data_channels_openfold = {"genetic": genetic_db, 'fasta': fasta, 'param': param}
estimator_openfold.fit(inputs=data_channels_openfold,
wait=False)To access the final output after the job completes, we run the following commands:
!aws s3 cp {estimator_openfold.model_data} openfold_output/model.tar.gz
!tar zxfv openfold_output/model.tar.gz -C openfold_output/Runtime performance
The following table shows the cost savings of 57% and 51% for AlphaFold and OpenFold, respectively, by splitting the MSA alignment and folding algorithms in two jobs as compared to a single compute job. It allows us to right-size the compute for each job: ml.m5.4xlarge for MSA alignment and ml.g5.2xlarge for AlphaFold and OpenFold.
| Job Details | Instance Type | Input FASTA Sequence | Runtime | Cost |
| MSA alignment + OpenFold | ml.g5.4xlarge | T1030 | 50 mins | $1.69 |
| MSA alignment + AlphaFold | ml.g5.4xlarge | T1030 | 65 mins | $2.19 |
| MSA alignment | ml.m5.4xlarge | T1030 | 46 mins | $0.71 |
| OpenFold | ml.g5.2xlarge | T1030 | 6 mins | $0.15 |
| AlphaFold | ml.g5.2xlarge | T1030 | 21 mins | $0.53 |
Build a repeatable workflow using SageMaker Pipelines
With SageMaker Pipelines, we can create an ML workflow that takes care of managing data between steps, orchestrating their runs, and logging. SageMaker Pipelines also provides us a UI to visualize our pipeline and easily run our ML workflow.
A pipeline is created by combing a number of steps. In this pipeline, we combine three training steps, which require an SageMaker estimator. The estimators defined in this notebook are very similar to those defined in 01-run_stepbystep.ipynb, with the exception that we use Amazon S3 locations to point to our inputs and outputs. The dynamic variables allow SageMaker Pipelines to run steps one after another and also permit the user to retry failed steps. The following screenshot shows a Directed Acyclic Graph (DAG), which provides information on the requirements for and relationships between each step of our pipeline.

Dynamic variables
SageMaker Pipelines is capable of taking user inputs at the start of every pipeline run. We define the following dynamic variables, which we would like to change during each experiment:
- FastaInputS3URI – Amazon S3 URI of the FASTA file uploaded via SDK, Boto3, or manually.
- FastFileName – Name of the FASTA file.
- db_preset – Selection between
full_dbsorreduced_dbs. - MaxTemplateDate – AlphaFold’s MSA step will search for the available templates before the date specified by this parameter.
- ModelPreset – Select between AlphaFold models including
monomer,monomer_casp14,monomer_ptm, andmultimer. - NumMultimerPredictionsPerModel – Number of seeds to run per model when using multimer system.
- InferenceInstanceType – Instance type to use for inference steps (both AlphaFold and OpenFold). The default value is ml.g5.2xlarge.
- MSAInstanceType – Instance type to use for MSA step. The default value is ml.m5.4xlarge.
See the following code:
fasta_file = ParameterString(name="FastaFileName")
fasta_input = ParameterString(name="FastaInputS3URI")
pipeline_db_preset = ParameterString(name="db_preset",
default_value='full_dbs',
enum_values=['full_dbs', 'reduced_dbs'])
max_template_date = ParameterString(name="MaxTemplateDate")
model_preset = ParameterString(name="ModelPreset")
num_multimer_predictions_per_model = ParameterString(name="NumMultimerPredictionsPerModel")
msa_instance_type = ParameterString(name="MSAInstanceType", default_value='ml.m5.4xlarge')
instance_type = ParameterString(name="InferenceInstanceType", default_value='ml.g5.2xlarge')A SageMaker pipeline is constructed by defining a series of steps and then chaining them together in a specific order where the output of a previous step becomes the input to the next step. Steps can be run in parallel and defined to have a dependency on a previous step. In this pipeline, we define an MSA step, which is the dependency for an AlphaFold inference step and OpenFold inference step that run in parallel. See the following code:
step_msa = TrainingStep(
name="RunMSA",
step_args=pipeline_msa_args,
)
step_alphafold = TrainingStep(
name="RunAlphaFold",
step_args=pipeline_alphafold_default_args,
)
step_alphafold.add_depends_on([step_msa])
step_openfold = TrainingStep(
name="RunOpenFold",
step_args=pipeline_openfold_args,
)
step_openfold.add_depends_on([step_msa]To put all the steps together, we call the Pipeline class and provide a pipeline name, pipeline input variables, and the individual steps:
pipeline_name = f"ProteinFoldWorkflow"
pipeline = Pipeline(
name=pipeline_name,
parameters=[
fasta_input,
instance_type,
msa_instance_type,
pipeline_db_preset
],
steps=[step_msa, step_alphafold, step_openfold],
)
pipeline.upsert(role_arn=role, # run this if it's the first time setting up the pipeline
description='Protein_Workflow_MSA')Run the pipeline
In the last cell of the notebook 02-define_pipeline.ipynb, we show how to run a pipeline using the SageMaker SDK. The dynamic variables we described earlier are provided as follows:
!mkdir ./sequence_input/
!curl 'https://www.predictioncenter.org/casp14/target.cgi?target=T1030&view=sequence' > ./sequence_input/T1030.fasta
fasta_file_name = 'T1030.fasta'
pathName = f'./sequence_input/{fasta_file_name}'
s3_fasta=sess.upload_data(path=pathName,
key_prefix='alphafoldv2/sequence_input')
PipelineParameters={
'FastaInputS3URI':s3_fasta,
'db_preset': 'full_dbs',
'FastaFileName': fasta_file_name,
'MaxTemplateDate': '2020-05-14',
'ModelPreset': 'monomer',
'NumMultimerPredictionsPerModel': '5',
'InferenceInstanceType':'ml.g5.2xlarge',
'MSAInstanceType':'ml.m5.4xlarge'
}
execution = pipeline.start(execution_display_name='SDK-Executetd',
execution_description='This pipeline was executed via SageMaker SDK',
parameters=PipelineParameters
)Track experiments and compare protein structures
For our experiment, we use an example protein sequence from the CASP14 competition, which provides an independent mechanism for the assessment of methods of protein structure modeling. The target T1030 is derived from the PDB 6P00 protein, and has 237 amino acids in the primary sequence. We run the SageMaker pipeline to predict the protein structure of this input sequence with both OpenFold and AlphaFold algorithms.
When the pipeline is complete, we download the predicted .pdb files from each folding job and visualize the structure in the notebook using py3Dmol, as in the notebook 04-compare_alphafold_openfold.ipynb.
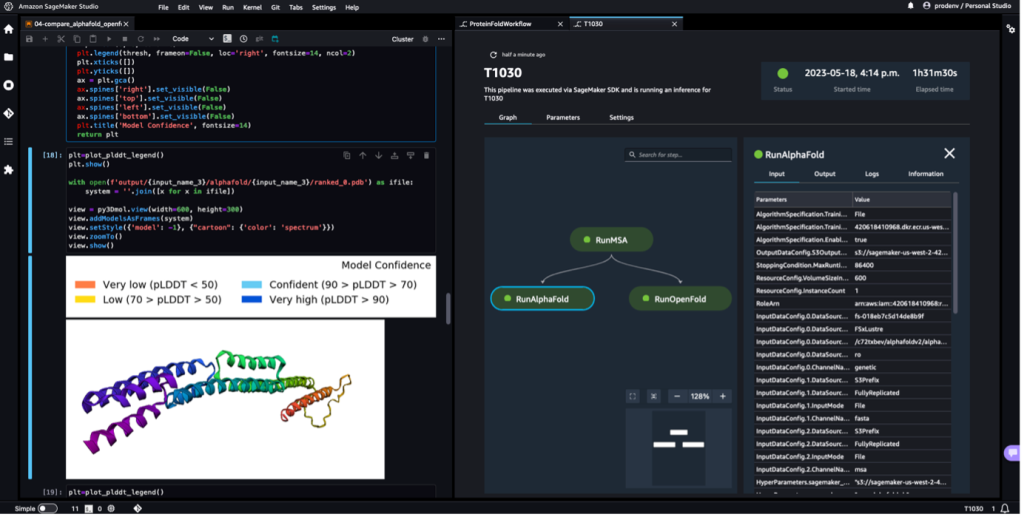
The following screenshot shows the prediction from the AlphaFold prediction job.

The predicted structure is compared against its known base reference structure with PDB code 6poo archived in RCSB. We analyze the prediction performance against the base PDB code 6poo with three metrics: RMSD, RMSD with superposition, and template modeling score, as described in Comparing structures.
| . | Input Sequence | Comparison With | RMSD | RMSD with Superposition | Template Modeling Score |
| AlphaFold | T1030 | 6poo | 247.26 | 3.87 | 0.3515 |
The folding algorithms are now compared against each other for multiple FASTA sequences: T1030, T1090, and T1076. New target sequences may not have the base pdb structure in reference databases and therefore it’s useful to compare the variability between folding algorithms.
| . | Input Sequence | Comparison With | RMSD | RMSD with Superposition | Template Modeling Score |
| AlphaFold | T1030 | OpenFold | 73.21 | 24.8 | 0.0018 |
| AlphaFold | T1076 | OpenFold | 38.71 | 28.87 | 0.0047 |
| AlphaFold | T1090 | OpenFold | 30.03 | 20.45 | 0.005 |
The following screenshot shows the runs of ProteinFoldWorkflow for the three FASTA input sequences with SageMaker Pipeline:

We also log the metrics with SageMaker Experiments as new runs of the same experiment created by the pipeline:
from sagemaker.experiments.run import Run, load_run
metric_type='compare:'
experiment_name = 'proteinfoldworkflow'
with Run(experiment_name=experiment_name, run_name=input_name_1, sagemaker_session=sess) as run:
run.log_metric(name=metric_type + "rmsd_cur", value=rmsd_cur_one, step=1)
run.log_metric(name=metric_type + "rmds_fit", value=rmsd_fit_one, step=1)
run.log_metric(name=metric_type + "tm_score", value=tmscore_one, step=1)We then analyze and visualize these runs on the Experiments page in SageMaker Studio.

The following chart depicts the RMSD value between AlphaFold and OpenFold for the three sequences: T1030, T1076, and T1090.

Conclusion
In this post, we described how you can use SageMaker Pipelines to set up and run protein folding workflows with two popular structure prediction algorithms: AlphaFold2 and OpenFold. We demonstrated a price performant solution architecture of multiple jobs that separates the compute requirements for MSA generation from structure prediction. We also highlighted how you can visualize, evaluate, and compare predicted 3D structures of proteins in SageMaker Studio.
To get started with protein folding workflows on SageMaker, refer to the sample code in the GitHub repo.
About the authors
 Michael Hsieh is a Principal AI/ML Specialist Solutions Architect. He works with HCLS customers to advance their ML journey with AWS technologies and his expertise in medical imaging. As a Seattle transplant, he loves exploring the great mother nature the city has to offer, such as the hiking trails, scenery kayaking in the SLU, and the sunset at Shilshole Bay.
Michael Hsieh is a Principal AI/ML Specialist Solutions Architect. He works with HCLS customers to advance their ML journey with AWS technologies and his expertise in medical imaging. As a Seattle transplant, he loves exploring the great mother nature the city has to offer, such as the hiking trails, scenery kayaking in the SLU, and the sunset at Shilshole Bay.
 Shivam Patel is a Solutions Architect at AWS. He comes from a background in R&D and combines this with his business knowledge to solve complex problems faced by his customers. Shivam is most passionate about workloads in machine learning, robotics, IoT, and high-performance computing.
Shivam Patel is a Solutions Architect at AWS. He comes from a background in R&D and combines this with his business knowledge to solve complex problems faced by his customers. Shivam is most passionate about workloads in machine learning, robotics, IoT, and high-performance computing.
 Hasan Poonawala is a Senior AI/ML Specialist Solutions Architect at AWS, Hasan helps customers design and deploy machine learning applications in production on AWS. He has over 12 years of work experience as a data scientist, machine learning practitioner, and software developer. In his spare time, Hasan loves to explore nature and spend time with friends and family.
Hasan Poonawala is a Senior AI/ML Specialist Solutions Architect at AWS, Hasan helps customers design and deploy machine learning applications in production on AWS. He has over 12 years of work experience as a data scientist, machine learning practitioner, and software developer. In his spare time, Hasan loves to explore nature and spend time with friends and family.
 Jasleen Grewal is a Senior Applied Scientist at Amazon Web Services, where she works with AWS customers to solve real world problems using machine learning, with special focus on precision medicine and genomics. She has a strong background in bioinformatics, oncology, and clinical genomics. She is passionate about using AI/ML and cloud services to improve patient care.
Jasleen Grewal is a Senior Applied Scientist at Amazon Web Services, where she works with AWS customers to solve real world problems using machine learning, with special focus on precision medicine and genomics. She has a strong background in bioinformatics, oncology, and clinical genomics. She is passionate about using AI/ML and cloud services to improve patient care.