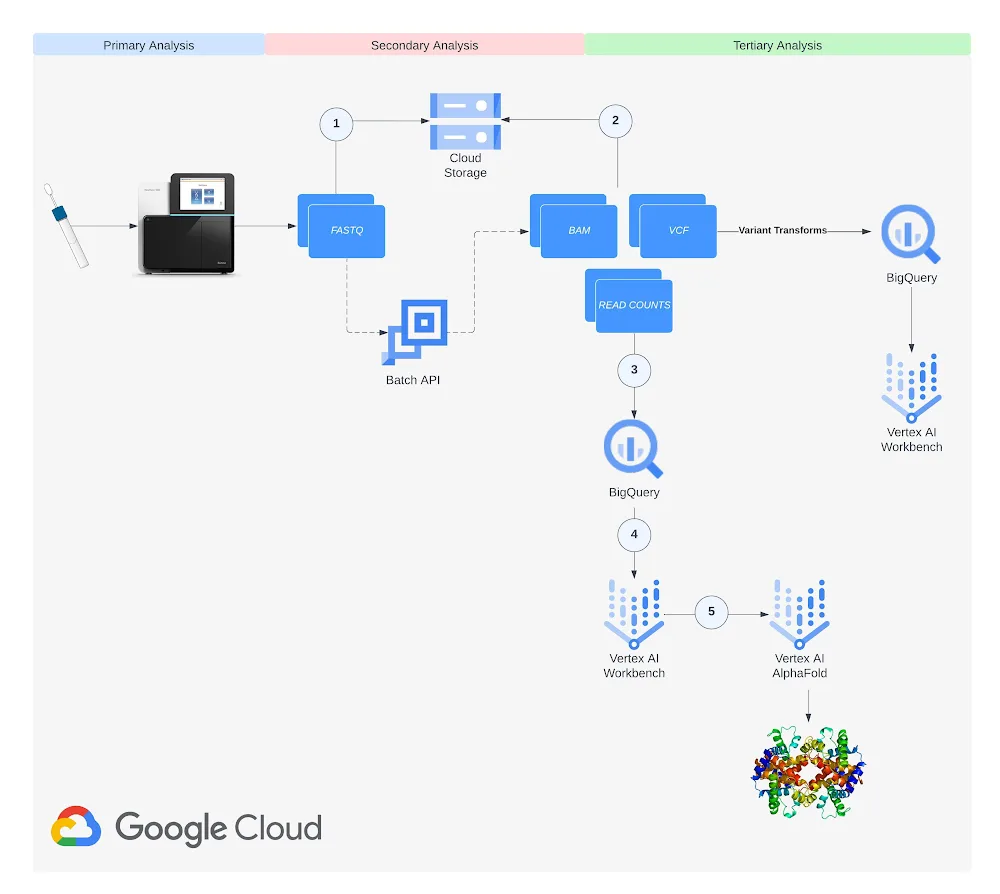
RNA-Seq and protein structure prediction are essential tools in modern biological research, facilitating insights into the molecular mechanisms of diseases and the development of potential therapies. RNA-Seq is a technique for profiling gene expression, enabling researchers to better understand gene regulation and complex interactions between genes. Protein structure prediction, on the other hand, provides information about a protein’s function and interactions with other molecules. This is useful in drug development and enables identification of target binding sites and optimization of drug candidates.
The challenge
The complexity of handling the large volumes of data generated by these techniques poses significant challenges for researchers. RNA-Seq and protein structure prediction require significant computational and storage overhead for accurate analysis. Protein structure prediction is further complicated by the size and complex binding dynamics of many proteins that make accurate prediction difficult. Additionally, the lack of ground truth structural data for many proteins makes it challenging to validate computational predictions, further adding to the complexity of the task. Addressing these challenges requires a scalable and reproducible analytical approach to achieve accurate and reliable results.
The solution
Recent advances in machine learning and cloud computing have opened up new avenues for tackling these challenges. AlphaFold, a deep learning program developed by DeepMind, can accurately predict a protein’s 3D structure and binding dynamics with other molecules from an input amino acid sequence. Nextflow, an open-source workflow management system, and Google Batch, a fully managed service to schedule, queue, and execute batch jobs on cloud computing resources, enable scalable and reproducible analysis of genomic data using Docker containers. Leveraging the power of Google Cloud, we have developed an end-to-end pipeline for RNA-Seq and protein structure prediction that utilizes BigQuery and Vertex AI to efficiently handle and process terabyte-scale data. By sharing our experience, we hope to provide insights into how Google Cloud can be used to tackle the computational challenges in modern biology and medicine, ultimately paving the way for new discoveries and innovations.
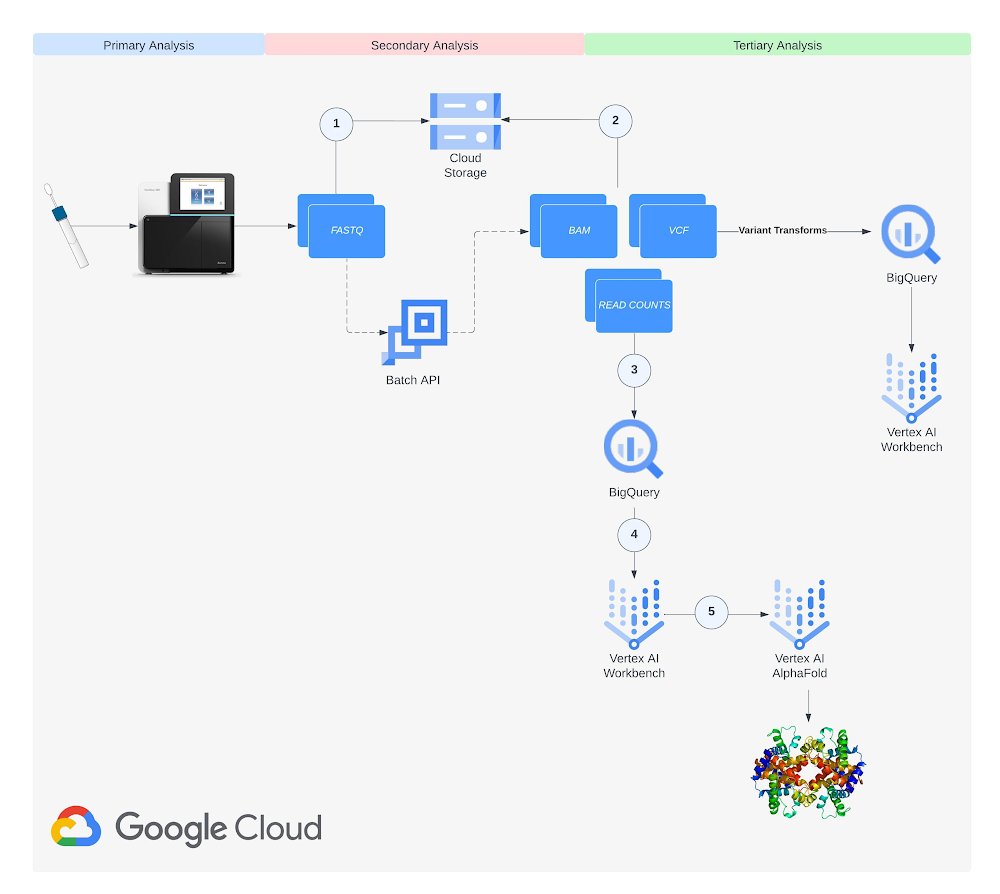
Methodology
Step 1: Setup
Create a Cloud Storage bucket. See documentation on creating a bucket. Enable the Artifact Registry, Batch, Dataflow, Cloud Life Sciences, Compute Engine, Notebooks, Cloud Storage, and Vertex AI APIs in your project. Create a service account and add the Cloud Life Sciences Workflows Runner, Service Account User, Service Usage Consumer, and Storage Object Admin roles at the project level. See documentation on creating a service account.
Step 2: Data collection
The next step in our solution involves data collection from a public NCBI dataset (GSE181830) sourced from a paper titled “Single-cell profiling of human bone marrow progenitors reveals mechanisms of failing erythropoiesis in Diamond-Blackfan anemia”. The patient metadata and FASTQ files (i.e. *.fq.gz) used in the study are downloadable via NCBI’s SRA Run Selector service and can be delivered directly to Cloud Storage by granting temporary Storage Object Admin access to NCBI’s service account on a specified bucket. Upon completion of data delivery, a confirmation email is sent, and the specified bucket should contain the files selected for delivery. Alternatively, you may download the FASTQ files from the European Nucleotide Archive (ENA) website by entering the SRA ID for each run and then using wget or curl to download files into a local directory and then copy them to a Cloud Storage Bucket using gsutil.
Step 3: Build genomic index
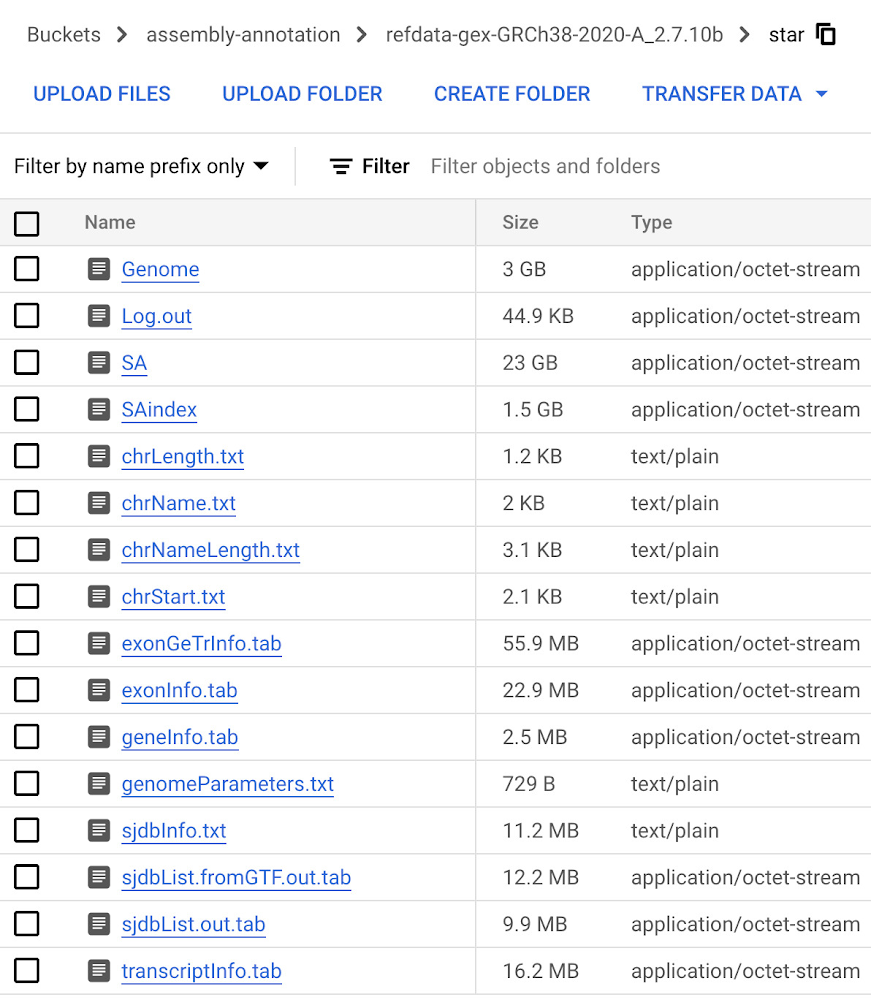
The next step involves the creation of a genome index (GRCh38.v42) that will be used to map reads against during the alignment step of our RNA-Seq analysis. Using transcript and annotation files downloaded from Gencode and uploaded to Cloud Storage, we build the genome index in a Vertex AI Workbench user-managed notebook using the open source STAR RNA aligner library (2.7.10c) available for download on Github. See documentation on creating a user-managed notebook instance. The process of creating the genome index with a specified output directory takes about an hour. Once finished, we upload the output directory to the Cloud Storage bucket created in Step 1.
Step 4: Build and upload a container image to Artifact Registry
The next step involves building and uploading a Docker container image to Artifact Registry, Google Cloud’s artifact repository. Nextflow executes each step in your pipeline in a container that you specify. We need to build the image that our pipeline will use and load it with dependencies and libraries including fastqc, multiqc, rna-star, rsem, and the BigQuery Python client library that will be referenced in our pipeline. See documentation on Creating a Repository and Pushing and Pulling images to Artifact Registry. Once the image has been created and pushed to a repository, grant the Artifact Registry Reader role to the service account created in Step 1. The Dockerfile we used in our workflow can be viewed in Github.
Step 5: Create BigQuery datasets for gene and isoform expression data
The next step is to create BigQuery datasets that will contain gene and isoform read count data respectively. The last step of the RNA-Seq pipeline writes read count data to BigQuery tables and the datasets must exist beforehand. See documentation on creating a dataset in BigQuery. Name the first created dataset rnaseq_nf_gene_results and the second rnaseq_nf_isoform_results. In both datasets, give the BigQuery Editor and BigQuery User roles to the service account created in Step 1.
Step 6: Configure a Nextflow pipeline
The next step is to configure a Nextflow pipeline to perform our paired-end RNA-Seq preprocessing workflow. This involves creating a nextflow.config and main.nf file in a Github repository. The nextflow.config, main.nf, and load_rsem_results_into_bq.py (referenced by main.nf) files we used in our workflow can be viewed in Github. See documentation on Nextflow scripting. Update the nextflow.config script with your project and bucket details. The steps of the pipeline detailed in the main.nf script are: (1) adapter and quality trimming with Trim Galore, (2) quality control readout with FastQC, (3) alignment and estimation of gene and isoform expression with RSEM, and (4) write of expression data to BigQuery.
Step 7: Deploy a pipeline on Nextflow Tower
The next step is to deploy a pipeline on Nextflow Tower. Tower provides support to authenticate to a Google project where the pipeline will run via a service account key. See documentation on connecting a service account key. Tower also provides support to connect to a Git repository that is hosting your Nextflow config using an access token. See documentation on connecting a repository in Tower. Once you have your service account and repository credentials configured, the next step is to create a compute environment. On the Add Compute Environment view, enter your name of choice for the environment (e.g. RNASeq-NF-GCP) and select Google Batch in the Platform drop down. Then, enter a region where the workload will be created (can use the same one specified in the nextflow.config from Step 6) and enter the pipeline work directory (i.e. GCS path).
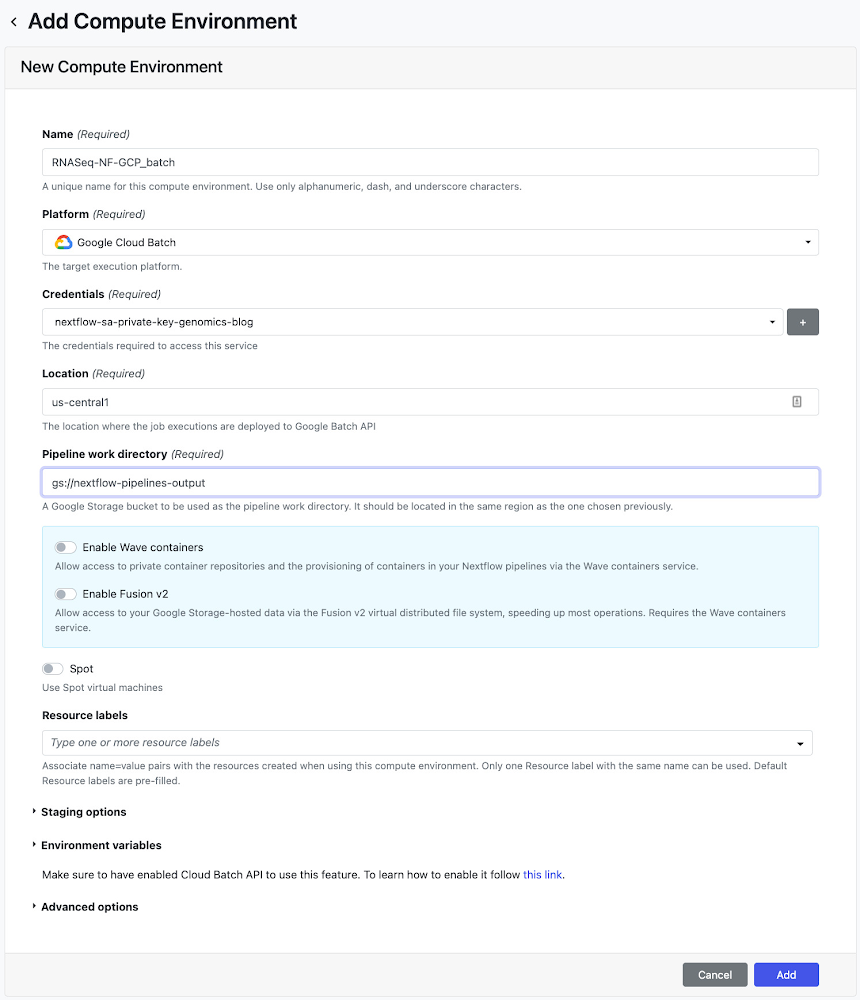
Once you have created the environment, go to the Add Pipeline view on the Launch Pad page. In the Add Pipeline view, enter your pipeline name of choice, select the compute environment created above, enter the Git repository URL where the Nextflow config created in Step 6 is hosted, and enter the revision number (i.e. branch) to use during execution. Lastly, enter paired-end-GRCh38 as the config profile as defined in the nextflow.config script.
Once you have created the pipeline, launch it. The pipeline should start in the background and take 4-6 hours. You can monitor the pipeline’s progress by going to the Runs tab and clicking the Run ID. Once the pipeline completes, genomic and isoform expression results will be available and queryable in the genomic and isoform tables created in Step 5. FastQC reports will be available in the output directory specified in the nextflow config.
Step 8: Run a DESeq2 notebook analysis
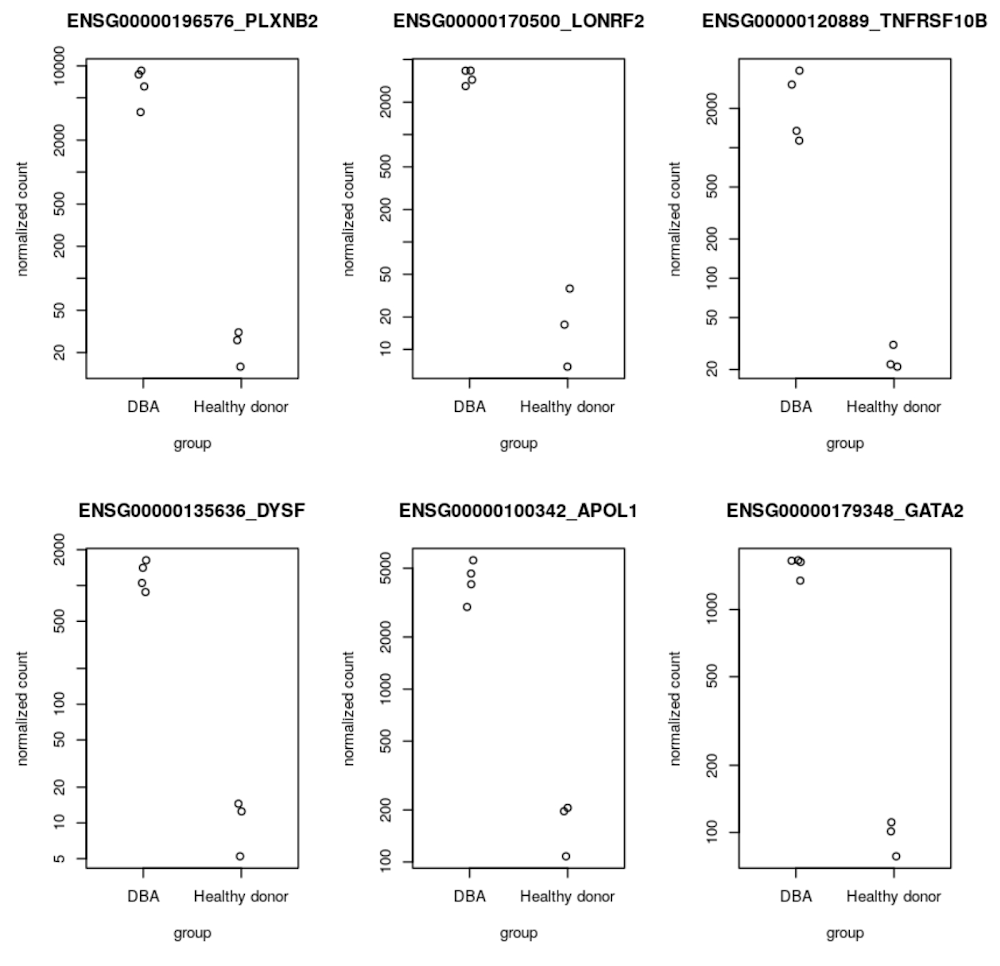
The next step is to run a DESeq2 analysis in a user-managed R notebook using the gene read expression data in BigQuery that was generated in Step 7. DESeq2 is an R package that provides statistical methods to detect differential gene expression between two or more biological conditions. In our workflow, we use DESeq2 to detect differentially expressed (DE) genes between patients with Diamond Blackfan anemia and healthy controls. We visualize a top DE gene in Step 10 using AlphaFold. See notebook in Github for reference. Several of the top DE genes output from the DESeq2 analysis, including GATA2, DYSF, and TNFRSF10B, are referenced in genomic literature as signatures of Diamond Blackfan anemia and validate the RNA-Seq pipeline preprocessing workflow and notebook analysis performed in Steps 1-8.
Step 9: Create remote function that converts transcript to amino acid sequence
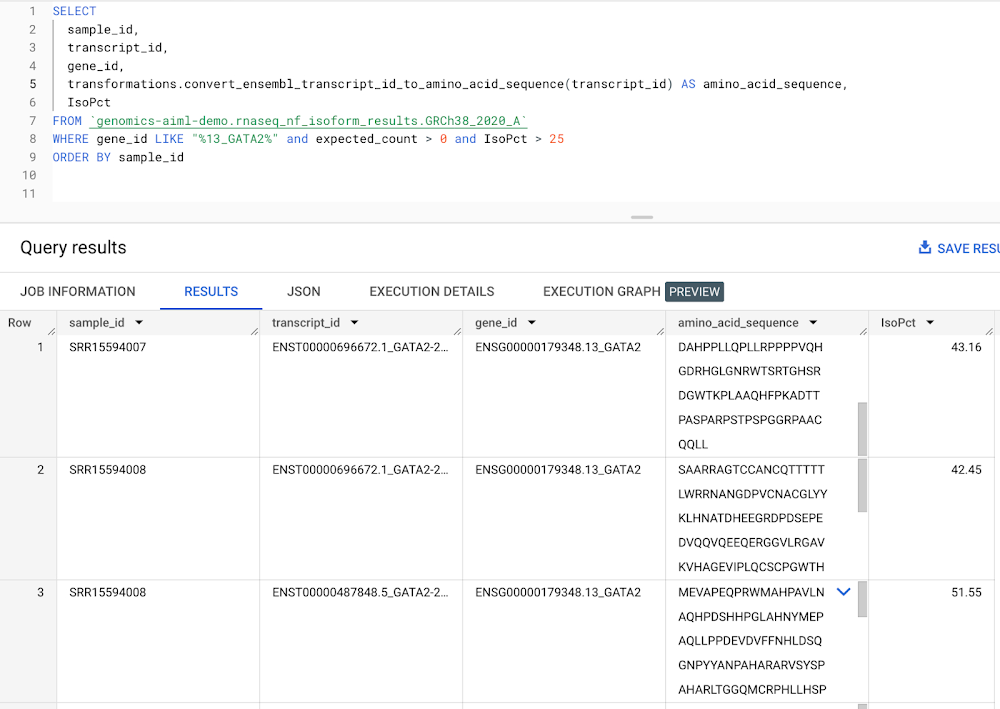
The next step is to create a remote function that converts a gene transcript to an amino acid sequence. The isoform expression table created in Step 7 includes a transcript ID column with isoform expression levels per gene. We pass the transcript ID column to a remote function that makes a call to the Ensembl Sequence REST API and outputs an amino acid sequence given an input gene transcript. Since AlphaFold accepts an amino acid sequence as input, this provides a scalable way to generate amino acid sequences that will be served to AlphaFold directly from expression data generated by our RNA-Seq pipeline. See documentation on creating a remote function. See code in Github for reference. In our workflow, we generate an amino acid sequence for transcripts corresponding to DE genes output in Step 8. An example query to create sequences for GATA2 transcripts is shown below.
Step 10: Visualize amino acid sequence with AlphaFold
The final step is to visualize an amino acid sequence with AlphaFold. Google Cloud provides a customized Docker image in Artifact Registry, with preinstalled packages, for starting a notebook instance in Vertex AI Workbench and running AlphaFold. You can find the notebook, the Dockerfile, and the build script in the Vertex AI community content. See related blog post for detailed instructions on deploying a notebook with recommended GPU accelerator. Using the BigQuery Python client, we perform a query to generate amino acid sequences for DE genes that are then passed and run through AlphaFold. An example protein structure prediction for one of the isoforms of the DE gene GATA2 is shown below.
Next steps
An expanded workflow would include implementation of a parallel variant calling pipeline on Nextflow, generation and upload of VCF files to Cloud Storage, and use of the Variant Transforms tool to load variant data into BigQuery. The isoform expression data from the RNA-Seq workflow would then be combined with the variant data to generate amino acids that are specific to each sample and contain the identified variants. This would provide additional support for a precision medicine-guided approach. See the variant-calling.nf script in Github for a reference variant calling pipeline.
In this blog, we have developed an end-to-end pipeline for RNA-Seq and protein structure prediction that utilizes BigQuery and Vertex AI in a scalable way to process genomic data. We hope we have provided insights into how Google Cloud can be used to tackle computational challenges in modern biology and medicine, paving the way for new discoveries and innovations.